Introduction
sideRETRO is a bioinformatic tool devoted for the detection of somatic (de novo) retrocopy insertion in whole genome and whole exome sequencing data (WGS, WES). The program has been written from scratch in C, and uses HTSlib and SQLite3 libraries, in order to manage SAM/BAM/CRAM reading and data analysis. The source code is distributed under the GNU General Public License.
Wait, what is retrocopy?
I can tell you now that retrocopy is a term used for the process resulting from reverse-transcription of a mature mRNA molecule into cDNA, and its insertion into a new position on the genome.
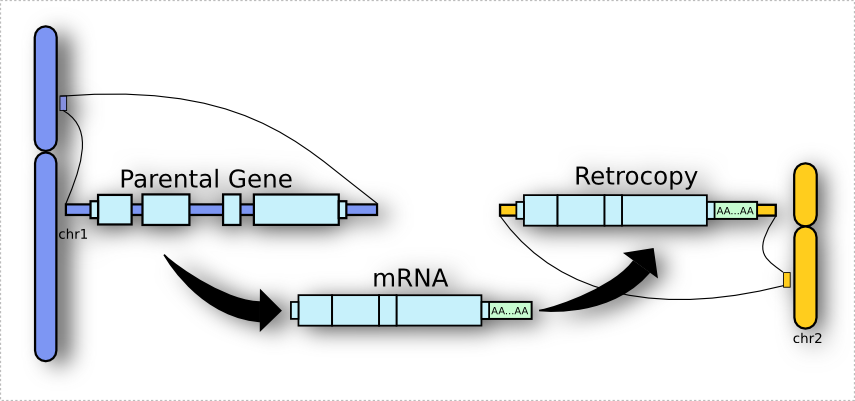
Got interested? For a more detailed explanation about what is a retrocopy at all, please see our section Retrocopy in a nutshell.
Features
When detecting retrocopy mobilization, sideRETRO can annotate several other features related to the event:
- Parental gene
The gene which underwent retrotransposition process, giving rise to the retrocopy.
- Genomic position
The genome coordinate where occurred the retrocopy integration (chromosome:start-end). It includes the insertion point.
- Strandness
Detects the orientation of the insertion (+/-). It takes into account the orientation of insertion, whether in the leading (+) or lagging (-) DNA strand.
- Genomic context
The retrocopy integration site context: If the retrotransposition event occurred at an intergenic or intragenic region - the latter can be splitted into exonic and intronic according to the host gene.
- Genotype
When multiple individuals are analysed, annotate the events for each one. That way, it is possible to distinguish if an event is exclusive or shared among the cohort.
- Haplotype
Our tool provides information about the ploidy of the event, i.e., whether it occurs in one or both homologous chromosomes (homozygous or heterozygous).
How it works
sideRETRO compiles to an executable called sider,
which has three subcommands: process-sample,
merge-call and make-vcf. The process-sample
subcommand reads a list of SAM/BAM/CRAM files, and captures
abnormal reads that must be related to an event of retrocopy.
All those data is saved to a SQLite3 database and then we come
to the second step merge-call, which processes the database
and annotate all the retrocopies found. Finally we can run the
subcommand make-vcf and generate an annotated retrocopy
VCF.
# List of BAM files
$ cat 'my-bam-list.txt'
/path/to/file1.bam
/path/to/file2.bam
/path/to/file3.bam
...
# Run process-sample step
$ sider process-sample \
--annotation-file='my-annotation.gtf' \
--input-file='my-bam-list.txt'
$ ls -1
my-genome.fa
my-annotation.gtf
my-bam-list.txt
out.db
# Run merge-call step
$ sider merge-call --in-place out.db
# Run make-vcf step
$ sider make-vcf \
--reference-file='my-genome.fa' out.db
Take a look at the manual page for installation and usage information. Also for more details about the algorithm, see our methodology.
Obtaining sideRETRO
The source code for the program can be obtaining in the github page. From the command line you can clone our repository:
$ git clone https://github.com/galantelab/sideRETRO.git
No Warranty
This program is distributed in the hope that it will be useful, but WITHOUT ANY WARRANTY; without even the implied warranty of MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the GNU General Public License for more details.
Reporting Bugs
If you find a bug, or have any issue, please inform us in the github issues tab. All bug reports should include:
The version number of sideRETRO
A description of the bug behavior
Citation
If sideRETRO was somehow useful in your research, please cite it:
@article{10.1093/bioinformatics/btaa689,
author = {Miller, Thiago L A and Orpinelli, Fernanda and Buzzo, José Leonel L and Galante, Pedro A F},
title = "{sideRETRO: a pipeline for identifying somatic and polymorphic insertions of processed pseudogenes or retrocopies}",
journal = {Bioinformatics},
year = {2020},
month = {07},
issn = {1367-4803},
doi = {10.1093/bioinformatics/btaa689},
url = {https://doi.org/10.1093/bioinformatics/btaa689},
note = {btaa689},
}
Further Information
If you need additional information, or a closer contact with the authors - we are always looking for coffee and good company - contact us by email, see authors.
Our bioinformatic group has a site, feel free to make us a visit: https://www.bioinfo.mochsl.org.br/.